
Hunting for huntingtin, part IV: What did you expect to find?
Hunting for ways to do the experiment
In our last episode, we were stopped in our tracks by some glutamines that were missing from our positive control. Sometimes it's hard enough to find the sequences we want; when the sequences are intentionally hidden, it's impossible.
We trudged onward, though, and identified the problem. All we had to do was to turn off the option to hide low complexity sequences and lo and behold, we found our glutamines ... at least we found them in a match to the positive control.
Next, I tried searching with a sequence of ten glutamines.
What did I get?
Nada.
Does that mean GenBank is devoid of proteins with 10 glutamines?
No. In this case, absence of evidence is not evidence of absence.
Huntingtin has at least 10 glutamines and sometimes more. Just because I didn't find a match, doesn't mean that there isn't a match to be found.
Hunting for ways around assumptions
This is a good time to talk about assumptions.
We've learned how to turn off the low complexity filter, but it looks like we will need to do a bit more. I've found this technique to be helpful before and we're going to have do it again. We're going to have to think like programmers in order to guess what assumptions were made in setting up the blastp web form at the NCBI server.
The most important assumption is given here:
It appears that the BLAST server forms at the NCBI were set up with the assumption that most people would use the collection of BLAST programs to look for homologous sequences.
Homologous protein or nucleic acid sequences are defined as sequences that share a common evolutionary origin. If two sequences are sufficiently similar, according to the statistical parameters in a BLAST search, they are likely to be homologous – that is they are likely to have been derived from a common ancestor. Naturally, the people who set up the web server at the NCBI made the assumption that this is reason why biologists would come to site to do BLAST searches.
There you have it.
The most commonly used set of bioinformatics programs, in the world, (outside of Microsoft Excel), work by calculating the probability that two sequences are related through evolution. (If you have problems with evolution, you probably want to look for another field. Take my advice; bioinformatics will not be a good fit.)
Because the BLAST web form was designed with the assumption that we're looking for statistically relevant homologous sequences, features like low complexity filtering are carried out by default. Low complexity sequences can be found in lots of proteins so their presence would not indicate a common evolutionary origin.
But that doesn't mean that we're not interested in finding them.
We have our own reasons for hunting, so what do we expect?
We are doing this search for an entirely different reason. We're not trying to use statistical significance to support the case that proteins with lots of glutamines share a common evolutionary ancestor. We're using BLAST as a surrogate for Google. We just want to find other proteins with polyglutamine sequences and see if those proteins share the characteristic, like huntingtin, that a genetic disease occurs when we have few extra glutamines.
Right, but how do we find those proteins with extra glutamines?
We go back to the blastp web form and look more closely at the default parameters.
Looking below the set of filters, we see that the default parameter for the Expect (E) value is set at 10. An E value of 10 means that we would expect to find 10 sequences that matched, if we searched a big enough database and it was filled with random sequences. When the cutoff is set to 10, any matching sequence with a value over 10 would be hidden.
Hunting with blastp and to heck with the statistics!
To heck with statistical relevance! I want my sequences!!!
I arbitrarily raised the setting on the E value to 50. A stretch of 10 glutamines is pretty short, and so we would expect to get a high E value even if we have perfect match. (A quick explanation of E values.)
This time, I got results. Over 3400 protein sequences matched my 10 glutamine query.
And check this out:

The best match has an E value of 33.
This explains why I didn't see any sequences match when the cut-off value was set at 10.
But how good a match was this, anyway?
Look at the results:
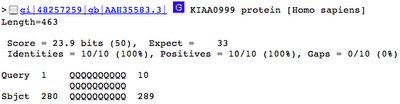
It was a perfect match.
Sometimes digital biology can be just like working in the lab. We spend lots of time just trying to get our controls to work.
Next time, we WILL do some experiments.
In the meantime, if you'd like to skip ahead, here's your assignment:
Find out if there are other cases where extra glutamines are linked to genetic disease.
And for extra credit - explain why this might be so. Use evidence to support your hypothesis.
Read the whole series:
Subject: Doing biology with bioinformatics
In our last episode, we were stopped in our tracks by some glutamines that were missing from our positive control. Sometimes it's hard enough to find the sequences we want; when the sequences are intentionally hidden, it's impossible.
We trudged onward, though, and identified the problem. All we had to do was to turn off the option to hide low complexity sequences and lo and behold, we found our glutamines ... at least we found them in a match to the positive control.
Next, I tried searching with a sequence of ten glutamines.
What did I get?
Nada.
Does that mean GenBank is devoid of proteins with 10 glutamines?
No. In this case, absence of evidence is not evidence of absence.
Huntingtin has at least 10 glutamines and sometimes more. Just because I didn't find a match, doesn't mean that there isn't a match to be found.
Hunting for ways around assumptions
This is a good time to talk about assumptions.
We've learned how to turn off the low complexity filter, but it looks like we will need to do a bit more. I've found this technique to be helpful before and we're going to have do it again. We're going to have to think like programmers in order to guess what assumptions were made in setting up the blastp web form at the NCBI server.
The most important assumption is given here:
To assess whether a given alignment constitutes evidence for homology, it helps to know how strong an alignment can be expected from chance alone.
From "The Statistics of Similarity Scores"
It appears that the BLAST server forms at the NCBI were set up with the assumption that most people would use the collection of BLAST programs to look for homologous sequences.
Homologous protein or nucleic acid sequences are defined as sequences that share a common evolutionary origin. If two sequences are sufficiently similar, according to the statistical parameters in a BLAST search, they are likely to be homologous – that is they are likely to have been derived from a common ancestor. Naturally, the people who set up the web server at the NCBI made the assumption that this is reason why biologists would come to site to do BLAST searches.
There you have it.
The most commonly used set of bioinformatics programs, in the world, (outside of Microsoft Excel), work by calculating the probability that two sequences are related through evolution. (If you have problems with evolution, you probably want to look for another field. Take my advice; bioinformatics will not be a good fit.)
Because the BLAST web form was designed with the assumption that we're looking for statistically relevant homologous sequences, features like low complexity filtering are carried out by default. Low complexity sequences can be found in lots of proteins so their presence would not indicate a common evolutionary origin.
But that doesn't mean that we're not interested in finding them.
We have our own reasons for hunting, so what do we expect?
We are doing this search for an entirely different reason. We're not trying to use statistical significance to support the case that proteins with lots of glutamines share a common evolutionary ancestor. We're using BLAST as a surrogate for Google. We just want to find other proteins with polyglutamine sequences and see if those proteins share the characteristic, like huntingtin, that a genetic disease occurs when we have few extra glutamines.
Right, but how do we find those proteins with extra glutamines?
We go back to the blastp web form and look more closely at the default parameters.
Looking below the set of filters, we see that the default parameter for the Expect (E) value is set at 10. An E value of 10 means that we would expect to find 10 sequences that matched, if we searched a big enough database and it was filled with random sequences. When the cutoff is set to 10, any matching sequence with a value over 10 would be hidden.
Hunting with blastp and to heck with the statistics!
To heck with statistical relevance! I want my sequences!!!
I arbitrarily raised the setting on the E value to 50. A stretch of 10 glutamines is pretty short, and so we would expect to get a high E value even if we have perfect match. (A quick explanation of E values.)
This time, I got results. Over 3400 protein sequences matched my 10 glutamine query.
And check this out:

The best match has an E value of 33.
This explains why I didn't see any sequences match when the cut-off value was set at 10.
But how good a match was this, anyway?
Look at the results:

It was a perfect match.
Sometimes digital biology can be just like working in the lab. We spend lots of time just trying to get our controls to work.
Next time, we WILL do some experiments.
In the meantime, if you'd like to skip ahead, here's your assignment:
Find out if there are other cases where extra glutamines are linked to genetic disease.
And for extra credit - explain why this might be so. Use evidence to support your hypothesis.
Read the whole series:
- Hunting for huntingtin, part I Background, reviews, biochemistry of glutamine, and a bit of comparative genomics
- Hunting for huntingtin, part II In which we're reminded that database searches are experiments, too.
- Hunting for huntingtin, part III Our continuing search for proteins with polyglutamine
- Hunting for huntingtin, part IV: What did you expect to find?
- Hunting for huntingtin, part V: BLASTing on forward
Subject: Doing biology with bioinformatics
posted by Sandra Porter at 2:52 PM
![]()
![]()









0 Comments:
Post a Comment
<< Home