
Hunting for huntingtin V: BLASTing on forward
This is the fifth article in our series on using digital biology to investigate Huntington's disease. Right now, since we know that extra glutamines are linked to Huntington's disease, we would like to know if other genetic diseases can result from extra glutamines.
If you wish to learn more about the story, the previous articles are described below. Or you can jump ahead and go straight to today's episode.
Enough of the recap, on with the experiment!
It took a few trys to figure out how to do the experiment. Now that we know how to do it, what should we do first?
We should learn a bit more about blastp. Nothing scary, mind you, but it's nice to have an idea how the information we put into a program is related to the output. So, before we go on, we're going to do a little experiment with blastp and see what happens when we search with different numbers of glutamines.
(We could also read about blastp, but hey, experimenting is more fun!)
Here is an image compiled from our colorful results:
Notice, how the length of our query sequence (lots of glutamines, abbreviated as Q) is related to color code. The color code, as shown in the color key above each alignment, is based on the alignment score. Scores in certain ranges are colored certain ways. You can see as the length of the match increases, the color changes, too. Our polyglutamine sequences give us a nice way to look at this relationship because we're using the same amino acid.
Why is that important? Keep reading and wonder no more!
I'll give you 10 points for a good match!
Did you ever think that calculating blastp alignment scores might be kind of like a game?
Indeed, it's a great game.
You get different amounts of points for matching different amino acids. And if your amino acids don't match, you get a penalty. The scoring table (also called a "matrix") is shown below. Click on the image to see it appear a bit larger, if you like.
I highlighted the value that's assigned when a glutamine (Q) matches another glutamine (Q). If one glutamine matches another, we get 5 points. Yeah!
Our final score is calculated by tallying up the points for each position in two aligned sequences. If we match 10 glutamines in a row, we get 50 points. We multiply by some mysterious factors (that may be discussed later) and finally, we end up with a score around 24.

Can I use my alignment points like frequent flyer miles? Who made the score sheet?
Why does matching another glutamine get 5 points and having a C (cysteine) match another cysteine get 9 points? Or when a W (tryptophan) matches another tryptophan, it gets 12 points?
Steve and Georgia Henikoff came up with these scores by looking at blocks of aligned sequences and determining how often a particular amino acid was replaced by a different amino acid (1). If the same amino acid was almost always found at the same position, it was assigned a higher score (i.e. W, tryptophan).
Notice, too, that our table has a row with a *. The * represents a position in a sequence where one amino acid is missing. Missing amino acids get assigned a negative score (-4) because they're are less common. This is probably because amino acid changes, in important regions, would harm the protein structure and impair the function.
We also get points if an amino acid is replaced by one with similar chemical properties. For example if we replace a lysine (K) with an arginine (R), we still get 2 points, because both amino acids have a positive charge. (If you want a key with chemical structures, or a key to the abbreviations, you can get them both here).
How do different numbers of glutamines affect the blastp results?
The graph below shows what happens when I do blastp searches with different numbers of glutamines. It only takes 10 glutamines to change the E value from 33 (not very significant) to 0.0001 (very significant).
The scores change a bit more slowly. I was surprised to see the relationship between the number of glutamines and the number of hits. Intuitively, it seemed to me that a smaller number of glutamines should match more things in the database. But the results contradict that notion.
I was surprised to see the relationship between the number of glutamines and the number of hits. Intuitively, it seemed to me that a smaller number of glutamines should match more things in the database. But the results contradict that notion.
Why did that happen?
When in doubt, look at the data
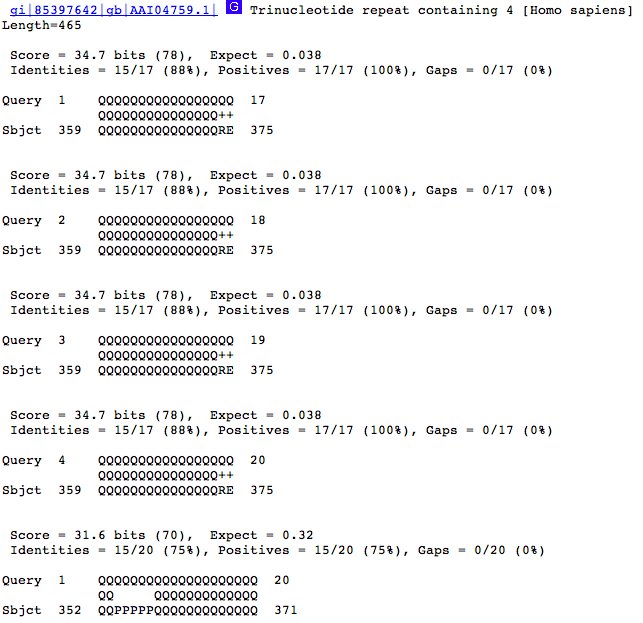
Looking at the sequence alignments shows why we see more hits. The results (below) show that when we we're using a repetitive sequence like QQQQQQ ... etc. and we use a longer query sequence, the query can start aligning to a database sequence at a greater number of points (and still have enough matching amino acids match to score as a hit). See how the alignment starts at 1, then 2, 3, and 4? This means that our query sequence can align to the same database sequence in multiple ways.
Longer query sequences can also tolerate gaps and more amino acid changes; yet still match enough amino acids to register as a hit.

So, in this case, a greater number of hits doesn't really mean that we're finding more sequences in the database. We're just finding the same sequence more times.
Do we only care about the number of hits?
No. Of course not. We do learn which proteins contain lots and lots of glutamines.
Stay tuned, next time we look at results.
References:
1. Henikoff, S. & Henikoff, J.G. (1992) "Amino acid substitution matrices from protein blocks." Proc. Natl. Acad. Sci. USA 89:10915-10919. (PubMed)
Subject: Doing biology with bioinformatics
If you wish to learn more about the story, the previous articles are described below. Or you can jump ahead and go straight to today's episode.
- Hunting for huntingtin, part I Background, reviews, biochemistry of glutamine, and a bit of comparative genomics
- We were quite busy in part I. We learned about Woody Guthrie, found an interview with Dr. Nancy Wexler, and got a bit of background information on the huntingtin gene.
- We used the UCSC genome browser to see if other creatures have the huntingtin gene and a similar gene structure.
- We asked if the extra glutamines caused the disease and looked at experiments where the Jackson Labs tested this idea in mice.
- Last we looked at the structure of glutamine and mused about why lots of glutamines might cause problems for a cell.
- Hunting for huntingtin, part II In which we're reminded that database searches are experiments, too.
- We discover that the polyglutamine regions are missing from structures with polyglutamine and we are unable to find polyglutamine sequences in GenBank
- Hunting for huntingtin, part III Our continuing search for proteins with polyglutamine
- In which we learn what happens to low complexity sequences in a blastp search.
- Hunting for huntingtin, part IV: What did you expect to find?
- In which we learn how to adjust the blastp parameters so that we can find proteins with polyglutamines
Enough of the recap, on with the experiment!
It took a few trys to figure out how to do the experiment. Now that we know how to do it, what should we do first?
We should learn a bit more about blastp. Nothing scary, mind you, but it's nice to have an idea how the information we put into a program is related to the output. So, before we go on, we're going to do a little experiment with blastp and see what happens when we search with different numbers of glutamines.
(We could also read about blastp, but hey, experimenting is more fun!)
Here is an image compiled from our colorful results:
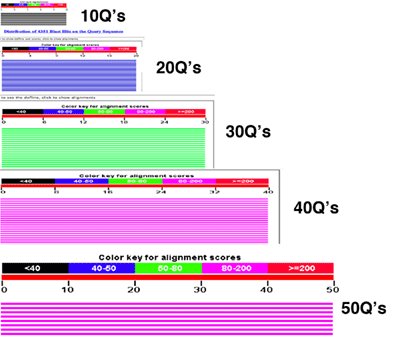
Notice, how the length of our query sequence (lots of glutamines, abbreviated as Q) is related to color code. The color code, as shown in the color key above each alignment, is based on the alignment score. Scores in certain ranges are colored certain ways. You can see as the length of the match increases, the color changes, too. Our polyglutamine sequences give us a nice way to look at this relationship because we're using the same amino acid.
Why is that important? Keep reading and wonder no more!
I'll give you 10 points for a good match!
Did you ever think that calculating blastp alignment scores might be kind of like a game?
Indeed, it's a great game.
You get different amounts of points for matching different amino acids. And if your amino acids don't match, you get a penalty. The scoring table (also called a "matrix") is shown below. Click on the image to see it appear a bit larger, if you like.
I highlighted the value that's assigned when a glutamine (Q) matches another glutamine (Q). If one glutamine matches another, we get 5 points. Yeah!
Our final score is calculated by tallying up the points for each position in two aligned sequences. If we match 10 glutamines in a row, we get 50 points. We multiply by some mysterious factors (that may be discussed later) and finally, we end up with a score around 24.

Can I use my alignment points like frequent flyer miles? Who made the score sheet?
Why does matching another glutamine get 5 points and having a C (cysteine) match another cysteine get 9 points? Or when a W (tryptophan) matches another tryptophan, it gets 12 points?
Steve and Georgia Henikoff came up with these scores by looking at blocks of aligned sequences and determining how often a particular amino acid was replaced by a different amino acid (1). If the same amino acid was almost always found at the same position, it was assigned a higher score (i.e. W, tryptophan).
Notice, too, that our table has a row with a *. The * represents a position in a sequence where one amino acid is missing. Missing amino acids get assigned a negative score (-4) because they're are less common. This is probably because amino acid changes, in important regions, would harm the protein structure and impair the function.
We also get points if an amino acid is replaced by one with similar chemical properties. For example if we replace a lysine (K) with an arginine (R), we still get 2 points, because both amino acids have a positive charge. (If you want a key with chemical structures, or a key to the abbreviations, you can get them both here).
How do different numbers of glutamines affect the blastp results?
The graph below shows what happens when I do blastp searches with different numbers of glutamines. It only takes 10 glutamines to change the E value from 33 (not very significant) to 0.0001 (very significant).
The scores change a bit more slowly.
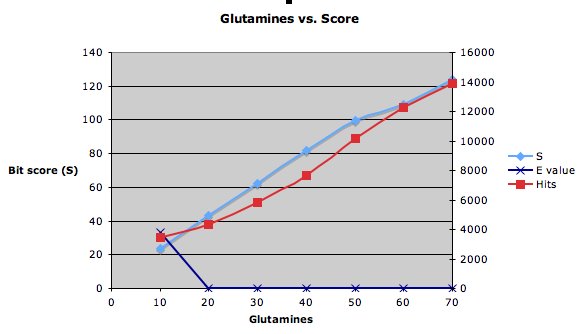 I was surprised to see the relationship between the number of glutamines and the number of hits. Intuitively, it seemed to me that a smaller number of glutamines should match more things in the database. But the results contradict that notion.
I was surprised to see the relationship between the number of glutamines and the number of hits. Intuitively, it seemed to me that a smaller number of glutamines should match more things in the database. But the results contradict that notion.Why did that happen?
When in doubt, look at the data
Looking at the sequence alignments shows why we see more hits. The results (below) show that when we we're using a repetitive sequence like QQQQQQ ... etc. and we use a longer query sequence, the query can start aligning to a database sequence at a greater number of points (and still have enough matching amino acids match to score as a hit). See how the alignment starts at 1, then 2, 3, and 4? This means that our query sequence can align to the same database sequence in multiple ways.
Longer query sequences can also tolerate gaps and more amino acid changes; yet still match enough amino acids to register as a hit.
So, in this case, a greater number of hits doesn't really mean that we're finding more sequences in the database. We're just finding the same sequence more times.
Do we only care about the number of hits?
No. Of course not. We do learn which proteins contain lots and lots of glutamines.
Stay tuned, next time we look at results.
References:
1. Henikoff, S. & Henikoff, J.G. (1992) "Amino acid substitution matrices from protein blocks." Proc. Natl. Acad. Sci. USA 89:10915-10919. (PubMed)
Subject: Doing biology with bioinformatics
technorati tags: biology, blast, , genetics, DNA
posted by Sandra Porter at 8:17 PM
![]()
![]()









0 Comments:
Post a Comment
<< Home